A. eMaize
请在quiz_emaize_tutorial_shared下载相关数据,并下载该文件夹下的内容,打开quiz_emaize_tutorial.ipynb文件阅读详细的Quiz指南。
eMaize背景简介
eMaize挑战赛是一个通过机器学习方法预测玉米性状的比赛,要求我们以SNP作为特征,通过训练一个模型,对玉米的三个性状进行预测。
本教程将会包括:
- 介绍数据的情况,使用方式
- 具体任务要求
- 一些机器学习的概念方法以及工具使用
编程工具介绍
大作业需要使用python完成,推荐读者使用python3。我们需要一些python的工具包来实现部分功能。推荐使用包管理软件Anaconda来预装一些必需的包以及安装其他需要的包。另外强烈建议使用jupyter notebook进行代码编辑、运行和调试。具体使用方法请参考教程Anaconda 和 jupyter相关指南。 如果本地缺少下列可能需要的包,请使用pip或者conda进行安装。如:
pip install tqdm
conda install sklearn
#导入必需的库
%pylab inline
import numpy as np
from sklearn.random_projection import SparseRandomProjection
import scipy.stats
import pandas as pd
import sklearn
import h5py
from sklearn.metrics import r2_score
from scipy.stats.stats import pearsonr
from sklearn.linear_model import LinearRegression
import xgboost
from xgboost.sklearn import XGBRegressor
from sklearn.linear_model import Ridge
from sklearn.kernel_ridge import KernelRidge
from sklearn import neighbors
from sklearn.ensemble import RandomForestRegressor
from sklearn.gaussian_process import GaussianProcessRegressor
from sklearn.gaussian_process.kernels import DotProduct
from tqdm import tqdm_notebook as tqdm
from IPython.display import display, Image
import seaborn as sns
import warnings
warnings.filterwarnings('ignore')
styles = ["white","dark",'whitegrid',"darkgrid"]
contexts = ['paper','talk','poster','notebook']
sns.set_context(contexts[1])
sns.set_style(styles[2])
数据介绍
原始数据中有6210个样本,其中4754个样本作为训练集,1456个样本作为测试集
genotype:SNP (Single-nucleotide polymorphism) 数据
每个位点可能有三种情况,如AA,AT,TT,每个样本SNP位点约为190万个,数据位置
data/genotype_2bit/,包含十个染色体各自的SNP,使用时需要整合在一起。trait:
共三种,trait1开花期,trait2株高,trait3产量,为连续值,只提供训练集样本的性状数据。
data/pheno_emaize.txt
作为示例仅仅使用每个样本的5000个SNP,在做大作业的过程中请使用全部的SNP。
Genotype数据
SNP数据存储格式
txt存储格式不适合大数据读取的问题,对内存的占用过多
对于结构化的、能够存储为矩阵的数据,可以使用HDF5格式存取,内存占用小,读取速度快。
tips: 在命令行查看数据shape的方法为: cd至文件路径下,输入:h5ls filename
#使用h5py读取5000个SNP,此时数据为原始的碱基信息
with h5py.File('data/snp_5000','r') as f:
print (list(f.keys()))
snps = f['snp'][:]
#使用h5py读取5000个SNP,此时为原始的碱基信息转化为数值信息后的数据
with h5py.File('data/2bit_geno','r') as f:
print (list(f.keys()))
snps_2bit = f['data'][:]
snps_2bit.shape
#使用h5py读取一个染色体的SNP示例,此时文件内为我们处理好的数值化数据,注意需要先解压文件:
with h5py.File('data/genotype_2bit/chr1.h5','r') as f:
print (list(f.keys()))
chr1_snps = f['data'][:]
chr1_snps.shape, chr1_snps.dtype
性状数据
使用numpy/pandas均可读取性状数据,计算时一般使用numpy.array的形式
前4764个样本的性状是已知的,后1454个样本性状待预测
traits = pd.read_csv('data/pheno_emaize.txt','\t')
display(traits.head())
display(traits.tail())
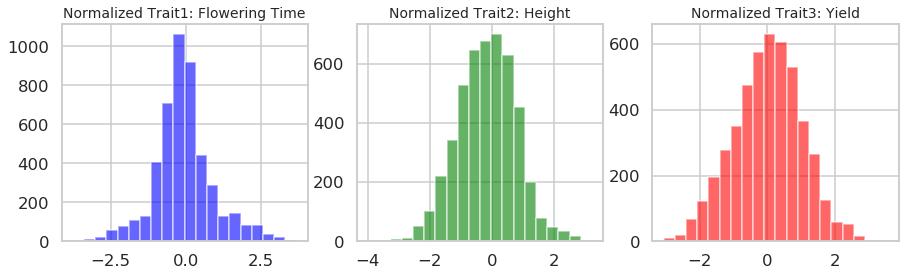
查看性状的分布情况
注意,对性状数据已经做过了normalization
trait1 = np.array(traits['trait1'])[:4754]
trait2 = np.array(traits['trait2'])[:4754]
trait3 = np.array(traits['trait3'])[:4754]
fig, ax = plt.subplots(1,3, figsize=(15,4))
ax[0].hist(trait1,bins = 20,color='b',alpha=0.6)
ax[0].set_title('Normalized Trait1: Flowering Time',fontsize=14)
ax[1].hist(trait2,bins = 20,color='g',alpha=0.6)
ax[1].set_title('Normalized Trait2: Height',fontsize=14)
ax[2].hist(trait3,bins = 20,color='r',alpha=0.6)
ax[2].set_title('Normalized Trait3: Yield',fontsize=14)
Quiz具体要求
之前的部分我们介绍了基本的背景知识,接下来我们会提出解答本题目的具体要求:
- 完成特征选择和特征筛除工作。
- 完成对三种性状的预测并提交预测结果,允许多次提交预测结果以获得更好的结果。
- 提交一份工作报告,中英文不限,同时提交源代码。
- 选择性完成加分项内容。
特征选择和冗余特征筛除
本挑战原始特征数量接近2,000,000,超过大多数机器学习模型的输入限制,特征间相关性很强,冗余特征很多,且过多的特征数量导致计算开销非常大,这都需要完成特征选择和去除冗余的步骤。
鉴于本问题原始特征数量过于巨大,并不是每种特征选择和降维方法都适合,请读者思考和选择合适的特征选择与降维方法,在这里仅提供几个方法参考:
- 特征选择:
- ANOVA(方差分析)
- 基于模型权重排序(如线性模型)
- 降维:
- PCA
- SVD
- Random Projection
tips: 请思考是否需要针对特征选择或者降维后的数据做scale
完成对测试集三种性状的预测,尝试得到尽可能好的预测结果
- 请思考和探索使用何种回归模型,读者可以尝试多种模型并比较其结果,在编程工具介绍部分读者可以看到一些机器学习模型的方法,也推荐读者思考和使用其他模型。
- 思考和探索是否对每个性状使用不同的模型
- 根据训练集与测试集的特殊划分方式(见查看训练集与测试集的划分部分)思考可以使用的策略。
加分内容
为了更完整地展示emaize挑战的困难与有趣之处,我们为有余力的读者设置了更多的挑战。
对模型进行鲁棒性测试 (10')
这是一个并不非常困难的但是对于本问题比较重要的工作,该工作可以细分为以下几项内容:
- 在读者已有的数据(训练集数据)上进行多轮(如100、1000轮)交叉验证,测试模型的鲁棒性
- 设计不同的数据集划分方式,除了随机划分训练集与验证集,还可以有其他特殊的设计方法
- 统计测试结果,以多种形式展现,包括统计数据和图示。
为了帮助读者完成测试,我们会给读者提供部分代码和一些相关的统计图,供读者使用和仿照设计,详见补充知识的模型鲁棒性测试部分。
使用ANOVA进行特征选择的加速算法 (5')
方差分析方法可以利用p值挑选feature
调用scipy.stats.f_oneway,利用SNPs和性状可以很容易地计算出p-value,进而挑选特征,但是对于大量数据来说速度较慢
我们可以设计一种加速ANOVA计算的方法完成计算,相比于scipy.stats的方法可以提升计算速度数百倍。为了帮助读者实现这一功能,我们提供给读者设计的基本思路,请参考ANOVA加速算法部分,有能力的读者可以根据基本思路实现ANOVA的加速算法。
混合线性模型 (20')
育种领域的一个经典模型是混合线性模型(linear mixed model),请尝试设计一个混合线性模型来解决本问题。
- 可以研究并调用FaST-LMM package,研究其原理并应用于我们的数据中,试图在测试集上获得好的预测结果(5')
- 根据其思路进行改进,设计一个类似的混合线性模型,并且可以通过快速的交叉验证挑选超参数,最终在测试集上获得好的效果。详细内容可以参考这篇文档。(15')
补充知识(选读)
测试预测结果
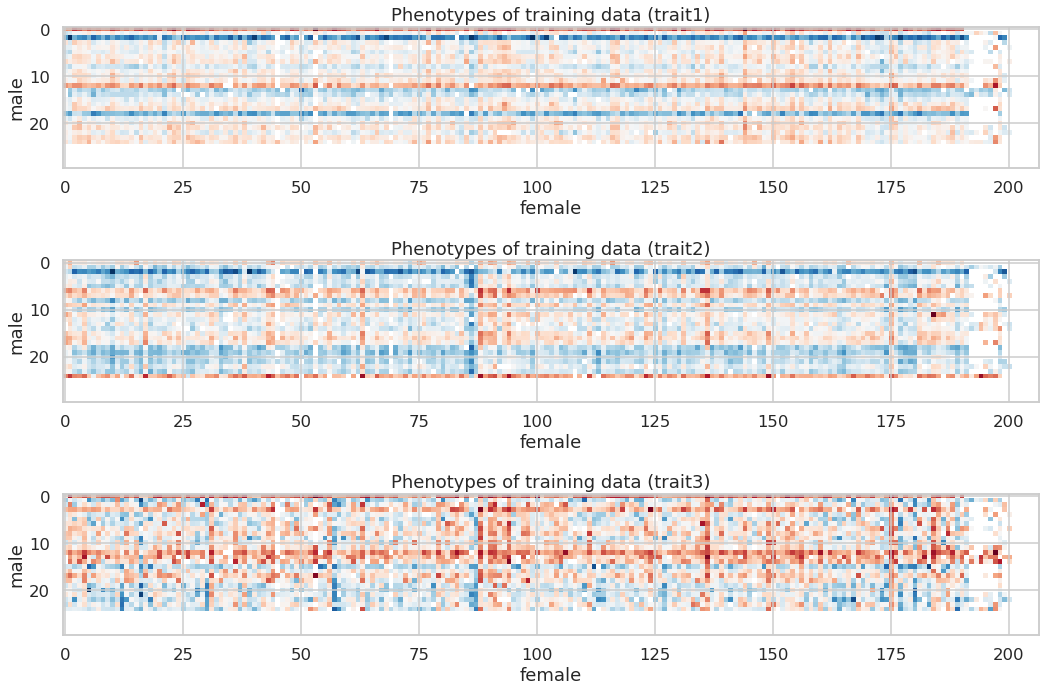
查看训练集与测试集的划分
请读者特别关注这个信息,训练集与测试集特殊的划分方式是本问题的一个特点与难点,也是想解决好本问题的关键。
下图中彩色部分为训练集性状,白色部分为待预测性状
可以发现其划分方式并不随机,这会导致常规的机器学习方法出现一些问题,常规的机器学习问题中,每个样本彼此独立,但是对于育种问题,很多样本可能有同一个父本或者母本,比如图中的每一行样本都来自同一个父本,每一列样本都来自同一个母本。读者需要自己思考待预测样本和已有样本的关系,结合机器学习的特点“通过学习已有数据的特征对未知数据进行预测”,思考并观察预测结果的好坏。
def generate_parent_table(phenotype_file):
phenotypes = pd.read_table(phenotype_file)
pedigree = phenotypes['pedigree'].str.split('_', expand=True)
pedigree.columns = ['f', 'X', 'm']
phenotypes = pd.concat([phenotypes, pedigree], axis=1)
phenotypes['number'] = np.arange(phenotypes.shape[0])
parent_table = phenotypes.pivot_table(values='number'
, index=['m'], columns=['f'], dropna=False)
male_ids = ['m%d' % i for i in range(1, parent_table.shape[0] + 1)]
female_ids = ['f%d' % i for i in range(1, parent_table.shape[1] + 1)]
parent_table = parent_table.loc[male_ids, female_ids]
return parent_table
phenotype_file = 'data/pheno_emaize.txt'
parent_table = generate_parent_table(phenotype_file)
phenotypes = pd.read_table('data/pheno_emaize.txt')
fig, ax = subplots(3,1, figsize=(15, 10))
for i in range(3):
trait = ['trait1','trait2','trait3'][i]
ax[i].imshow(np.take(np.ravel(phenotypes[trait].values), parent_table), cmap=cm.RdBu)
ax[i].set_title('Phenotypes of training data (%s)'%trait)
ax[i].set_ylabel('male')
ax[i].set_xlabel('female')
plt.tight_layout()
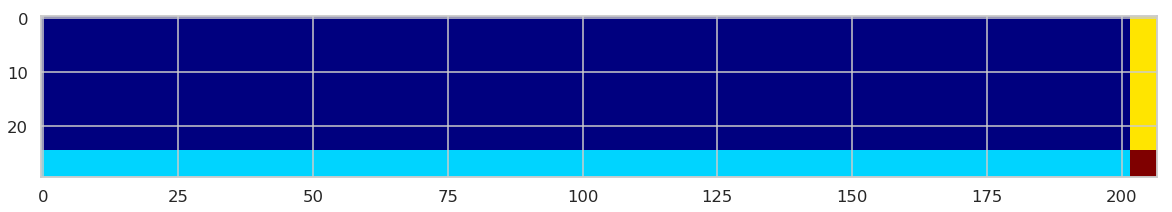
测试集具体划分
我们会把测试区域分为三个部分,来测试提交的结果,三个区域分别为下图的淡蓝色,黄色和红色区域,请读者思考它们各自的特征。
fig,ax=plt.subplots(figsize=(20,5))
testexample = np.zeros([30,207])
testexample[25:,:202] = 100
testexample[:25,202:] = 200
testexample[25:,202:] = 300
ax.imshow(testexample,cmap='jet')
评价指标
对于回归问题,我们一般使用和pearson correlation coefficient(PCC)衡量,其定义如下:
将SNP数据编码为向量
每个位点的碱基只有三种情况,不会出现更多碱基组合的可能,比如某位点仅有AA,AT,TT三种可能的情况
我们可以采取三种方式对其编码:
- 转化为0、1、2。找到minor allele frequency(MAF),即两种碱基(如A、T)中出现频率低的那个,以A作为MAF为例,则TT为0,AT为1,AA为2,这样可以突出MAF
- 转化为3-bit one hot vector,这样可以保持三种向量在空间距离的一致
转化为2-bit vector,则AA,AT,TT分别编为,不需要考虑MAF
我们采取第三种方式处理了数据并提供给读者
具体处理过程
def convert_2bit(seq):
genotypes = np.zeros([6210,2])
a = seq[1].split('/')
for i in range(6210):
if seq[4:][i] == a[0] + a[0]:
genotypes[i] = np.array([0,1])
if seq[4:][i] == a[0] + a[1]:
genotypes[i] = np.array([1,0])
if seq[4:][i] == a[1] + a[1]:
genotypes[i] = np.array([1,1])
genotypes = genotypes.astype('int').T
return genotypes
真实转换时python的转换速度较慢,这里为了展示基本的原理使用了python来演示。
snps = snps.astype('str')
snps.shape
convert_2bit(snps[2].astype('str'))
geno_conv = np.ndarray([10000,6210])
for i in tqdm(range(5000)):
geno_conv[i*2:(i+1)*2] = convert_2bit(snps[i+1])
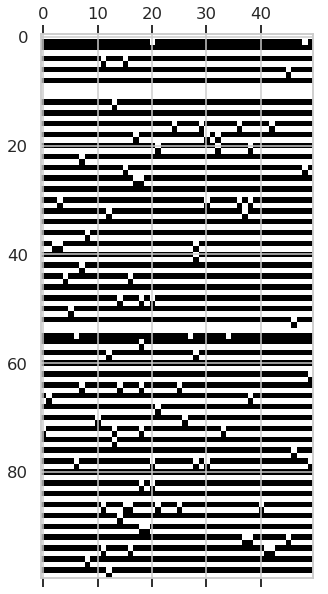
#查看SNP的大致情况,取前100行(50个位点),前50个样本,
fig, ax = plt.subplots(figsize=(20,10))
ax.matshow(geno_conv[:100,:50],cmap = cm.binary_r)
模型鲁棒性测试
设计特殊的交叉验证方式
不同的样本具有关联性,有的样本可能来自同一亲本,而训练集和测试集的划分并不是随机的 因此我们可以专门设计交叉验证时数据集的划分方式:
- random
- by female
- by male
- cross sampling
for method in ('random', 'by_female', 'by_male', 'cross'):
with h5py.File('data/cv_index.%s'%method, 'r') as f:
index_train = f['0/train'][:]
index_test = f['0/test'][:]
fig, ax = subplots(2, 1, figsize=(16, 6))
sampling_table = np.zeros(np.prod(parent_table.shape))
sampling_table[index_train] = 1
sampling_table = np.take(sampling_table, parent_table)
ax[0].imshow(sampling_table, cmap=cm.Greys)
ax[0].set_title('Training samples (%s)'%method)
sampling_table = np.zeros(np.prod(parent_table.shape))
sampling_table[index_test] = 1
sampling_table = np.take(sampling_table, parent_table)
ax[1].imshow(sampling_table, cmap=cm.Greys)
ax[1].set_title('Test samples (%s)'%method)
plt.tight_layout()
请在jupyter文件中查看相关图片。
ANOVA加速算法
思路简要提示:
