4.3 GSEA
1) Pipeline
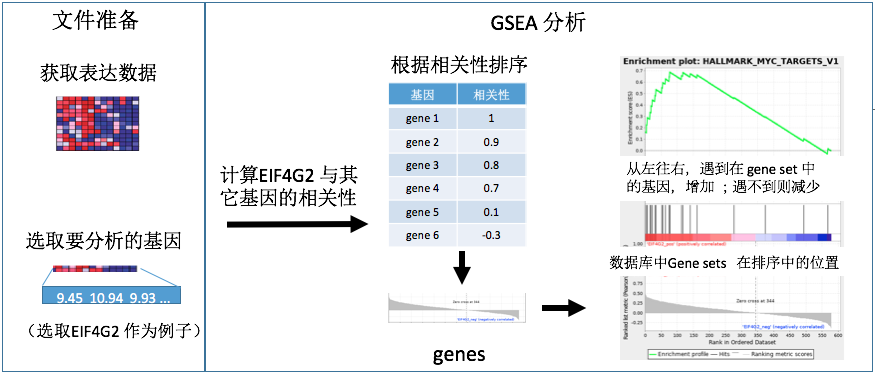
2) data structure
2a) getting software & data
- 程序下载与安装(Docker 中均已安装好):
- 进入 GSEA官方下载页面(需要登录方可进入,只需输入邮箱即可),在
Softwaretab下载 "javaGSEA Java Jar file"。已经安装。 - 安装 Java8 。已经安装
- qGSEA 已经安装。感兴趣请看 4) Tips/Utilities 安装 qGSEA。
- 进入 GSEA官方下载页面(需要登录方可进入,只需输入邮箱即可),在
- 准备数据:至 GEO 查找感兴趣的数据集(本例中我们使用 GSE19161),下载
series_matrix.txt.gz文件和family.soft.gz文件(Docker 中已经下载好)。
2b) input format
| Format | Description | Notes |
|---|---|---|
| txt, res, gct, pcl | A dataset containing expression value for each feature in each sample. | 658个基因的表达值(所有基因的表达值) |
| cls | Associates each sample with a phenotype label. | 基因在不同样品下的表达值 |
| Chip | Lists each probe and its matching HUGO gene symbol. | 基因 ID 和 基因名字对应表 |
| gmx or gmt | gives the gene set name and list of features in it. | 特定功能的基因集合 |
参见 http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm#_Preparing_Data_Files
2c) output format
网页格式的输出信息,使用说明见 http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm#_Interpreting_GSEA_Results。
3) Running Steps
这里我们分析 GSE19161 数据集中与 EIF4G2 基因的表达量显著相关的 gene set。
首先进入到容器(在自己电脑的 Terminal 中运行,详情请参见 这里):
docker exec -it bioinfo_tsinghua bash
以下步骤均在 /home/test/gsea/ 下进行:
cd /home/test/gsea/
3a) 准备 GSEA 输入文件
官方指南见 http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/Data_formats
如果你想利用 GEO 的原始数据,qGSEA 这个R包会让你轻松许多。
- 我们已经在
raw中下载好了原始数据文件:- matrix 文件
GSE19161_series_matrix.txt.gz - SOFT 文件
GSE19161_family.soft.gz
- matrix 文件
- 生成 GSEA 输入文件(打开R)
R
# 进入 R 操作界面
> dir.create('input/')
> qGSEA::make_gsea_input(
matrix_file = 'raw/GSE19161_series_matrix.txt.gz',
soft_file = 'raw/GSE19161_family.soft.gz',
output_dir = 'input/',
gene = 'EIF4G2'
)
> q()
Save workspace image? [y/n/c]: n # 按 n 再按 Enter
现在目录结构如下:
gsea ├── input │ ├── GSE19161.chip │ ├── GSE19161.cls │ └── GSE19161.txt └── raw ├── GSE19161_family.soft.gz └── GSE19161_series_matrix.txt.gz
3b) 在 command line 下运行GSEA
GSEA 可以以图形化界面或 command line 模式运行。由于 Docker 的限制,我们将前者移到了 这里。至于 command line 模式,官方指南 并没有对参数的名称和意义给出详细的解释,而是推荐在图形化界面中运行成功后,导出其所用的命令(示例见 这里)。
这里我们已经事先从图形化界面中导出了所需的命令:
java -cp /usr/local/GSEA/gsea-3.0.jar -Xmx512m xtools.gsea.Gsea -res input/GSE19161.txt -cls input/GSE19161.cls#EIF4G2 -chip input/GSE19161.chip -out output -gmx gseaftp.broadinstitute.org://pub/gsea/gene_sets_final/h.all.v6.2.symbols.gmt -metric Pearson
# java -cp /usr/local/GSEA/gsea-3.0.jar -Xmx512m xtools.gsea.Gsea \
# -res input/GSE19161.txt -cls input/GSE19161.cls#EIF4G2 -chip input/GSE19161.chip -out output \
# -gmx gseaftp.broadinstitute.org://pub/gsea/gene_sets_final/h.all.v6.2.symbols.gmt -metric Pearson
- 第一行指定运行的程序
- 第二行是输入/输出文件的路径(其中
#EIF4G2指定了我们要分析的基因) - 第三行设置了一些参数:
- 这里我们使用 hallmark 这个 gene set collection (
h.all.v6.1.symbols.gmt); - 另外,由于我们使用的是连续性表型(EIF4G2 基因的表达量),Metric 要选
Pearson
- 这里我们使用 hallmark 这个 gene set collection (
输出结果为 output/my_analysis.Gsea.1534401449035/ (最后的数字每次运行都不尽相同)。可以将其复制到 /home/test/share 中,然后在自己的电脑(或虚拟机)中用浏览器打开其中的 index.html,示例可在这里查看。
其中 my_analysis 可以用 -rpt_label 指定为其它值,如 -rpt_label GSE19161。但GSEA会自动在其后添加 .Gsea.1534401449035 来组成输出文件夹名。
4) Tips/Utilities
选择性练习
- 安装 qGSEA(打开R)
R
# 进入 R 操作界面
> if (!('devtools' %in% .packages(T))) install.packages('devtools')
> devtools::install_github('dongzhuoer/qGSEA')
> q()
Save workspace image? [y/n/c]: n # 按 n 再按 Enter。
4a) 使用 qGSEA 下载 GEO 的原始数据
新建一个空文件夹,进入其中,打开R
R
# 进入 R 操作界面
> dir.create('raw/')
> download.file(rGEO::gse_soft_ftp('GSE19161'), 'raw/GSE19161_family.soft.gz')
> download.file(rGEO::gse_matrix_ftp('GSE19161'), 'raw/GSE19161_series_matrix.txt.gz')
> q()
Save workspace image? [y/n/c]: n # 按 n 再按 Enter, 不保存 R 的修改。
/home/test/gsea/raw 中的原始数据文件即是用上述代码得到的。
4b) 以图形化界面运行 GSEA
由于 Docker 中无法启动图形化界面,本步骤需要在自己的电脑或虚拟机中进行。
- 切换到自己的电脑,装好全部软件,新建一个空文件夹
gsea/,在其中准备好输入文件。 打开 GSEA:
java -jar /path/to/gsea-x.y.jar,用文件管理器打开input/文件夹,将上述生成的文件拖拽到指定区域。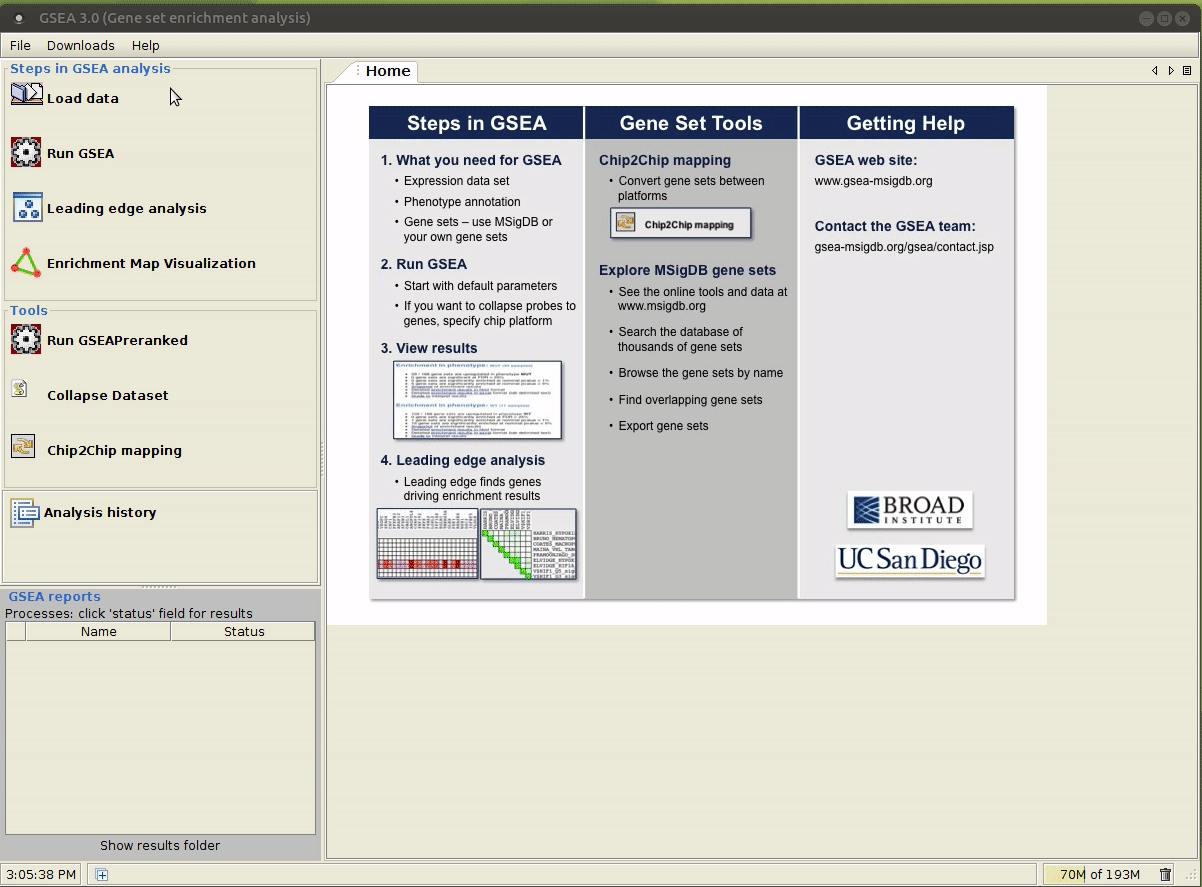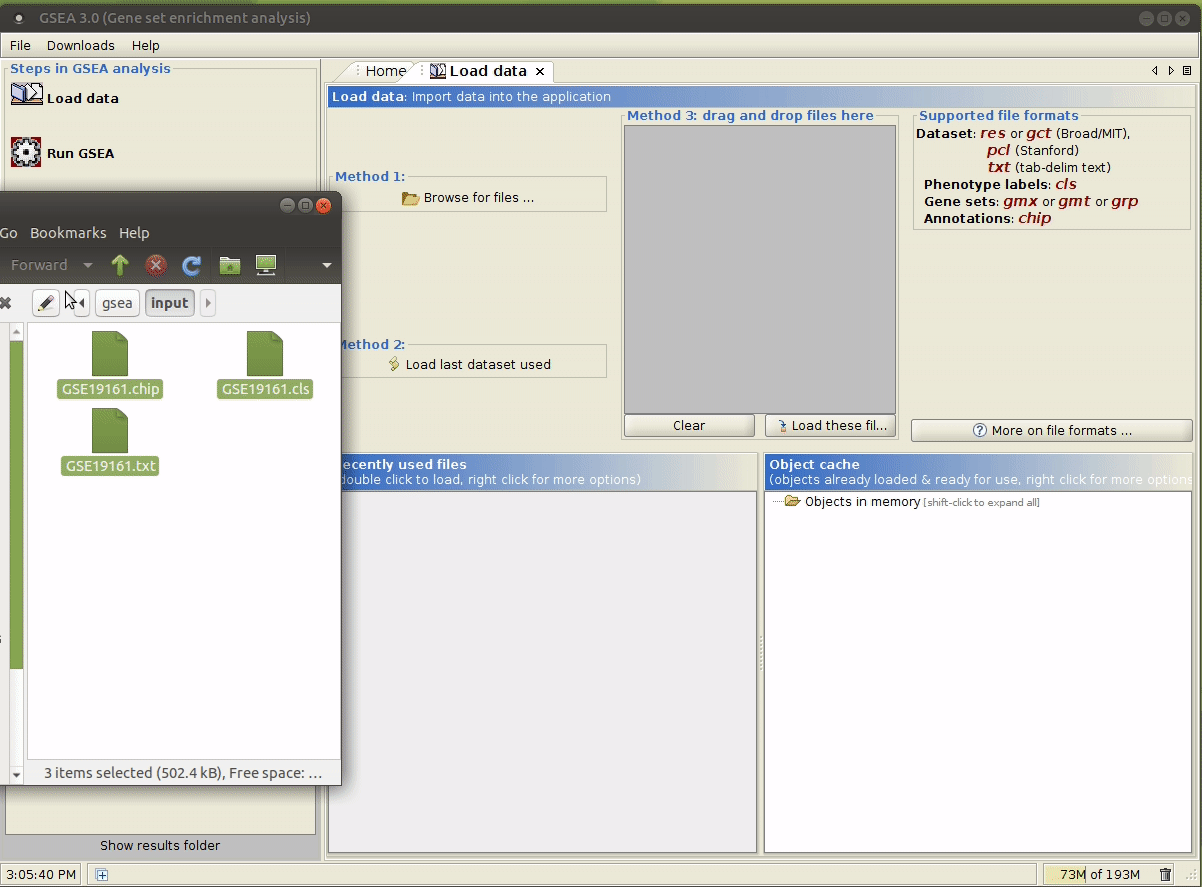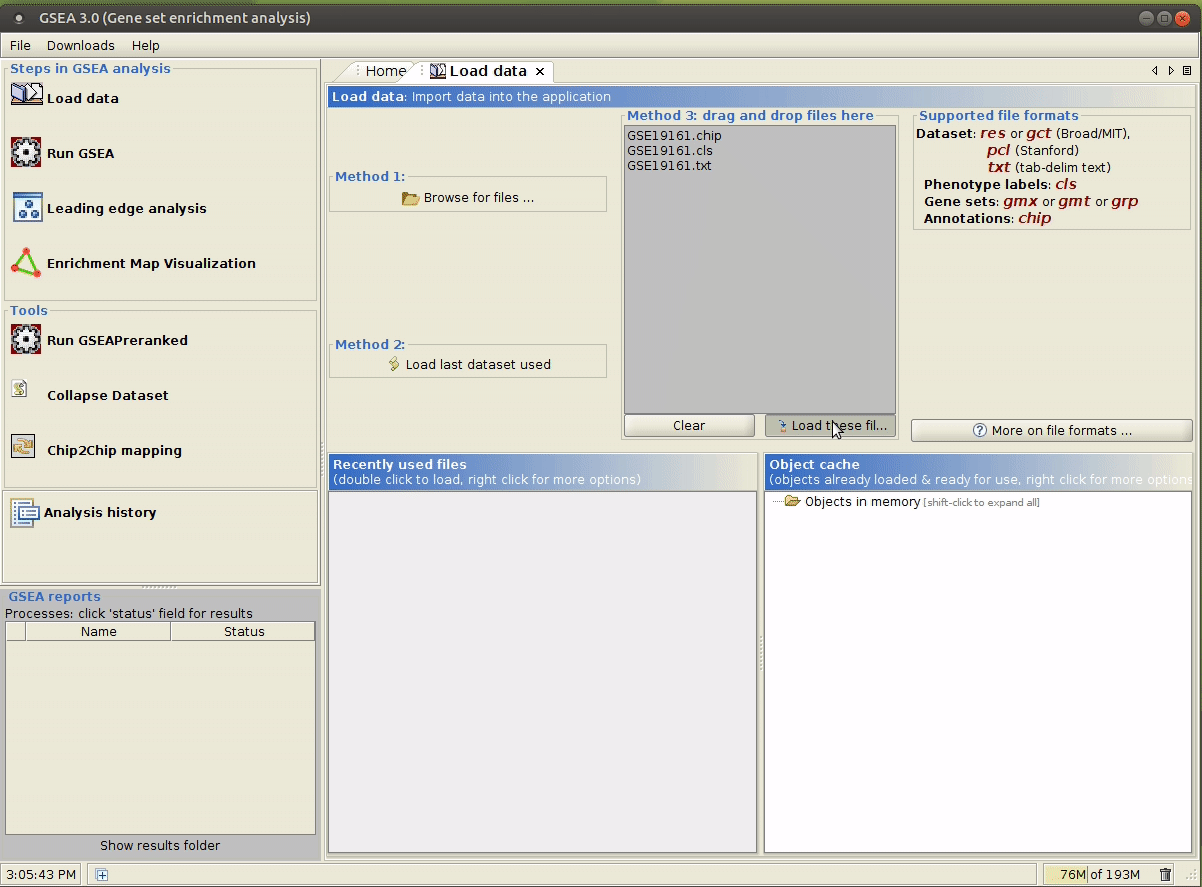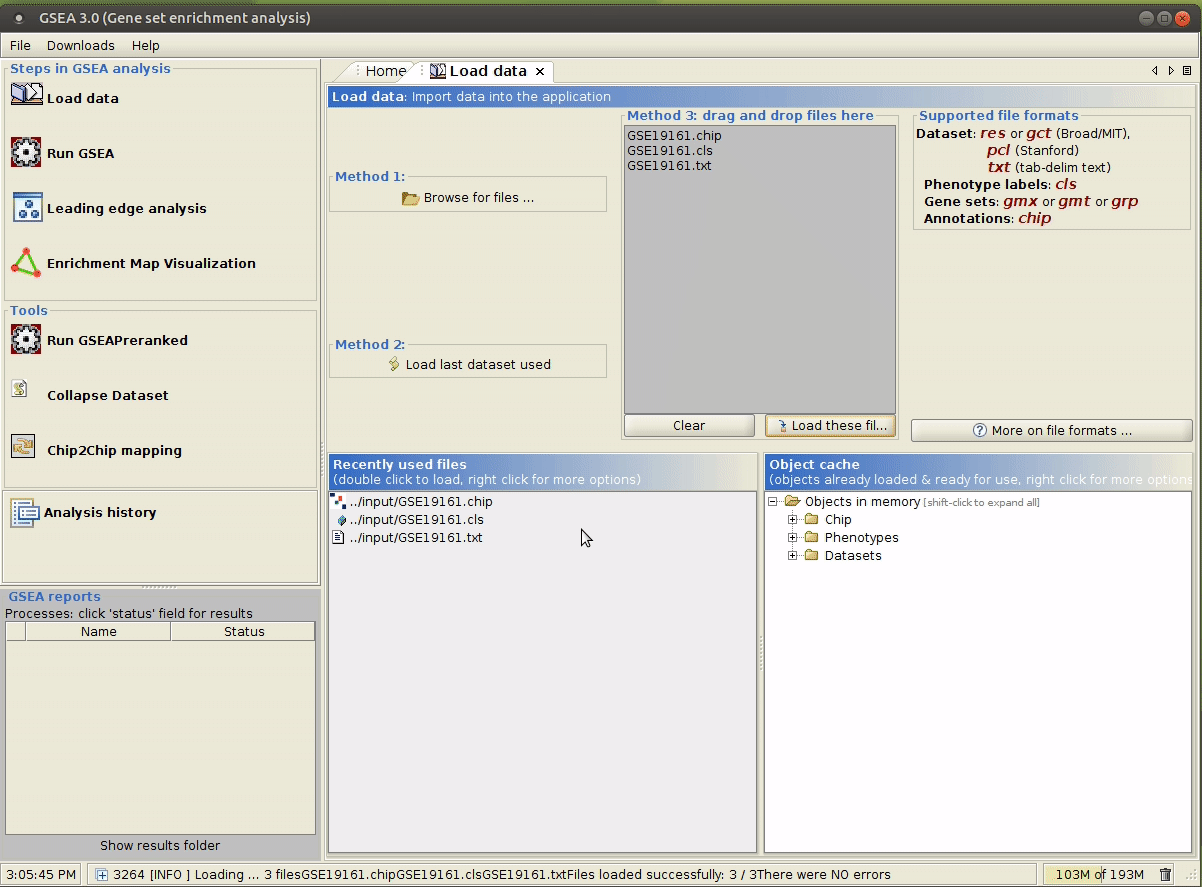
选择合适的参数,参数意义详见 command line 模式 第 3 行的解释。
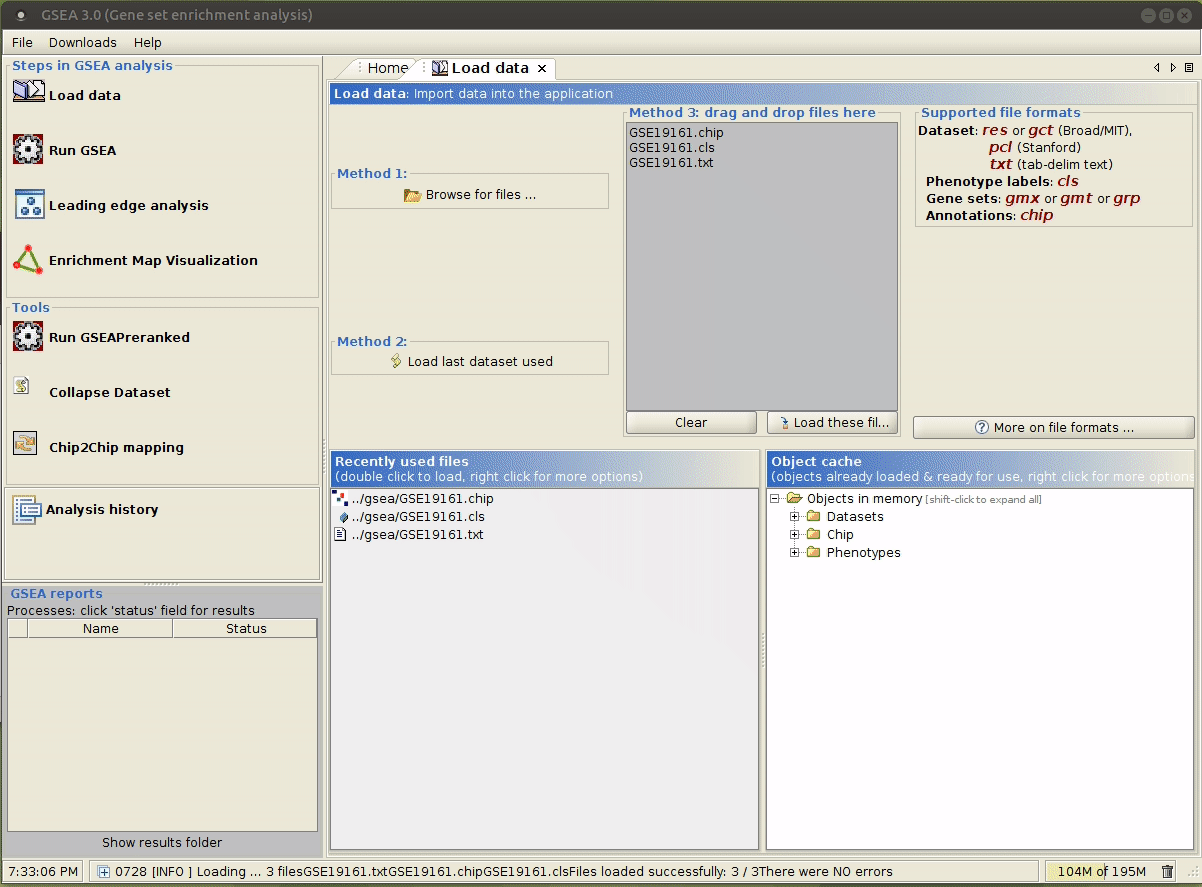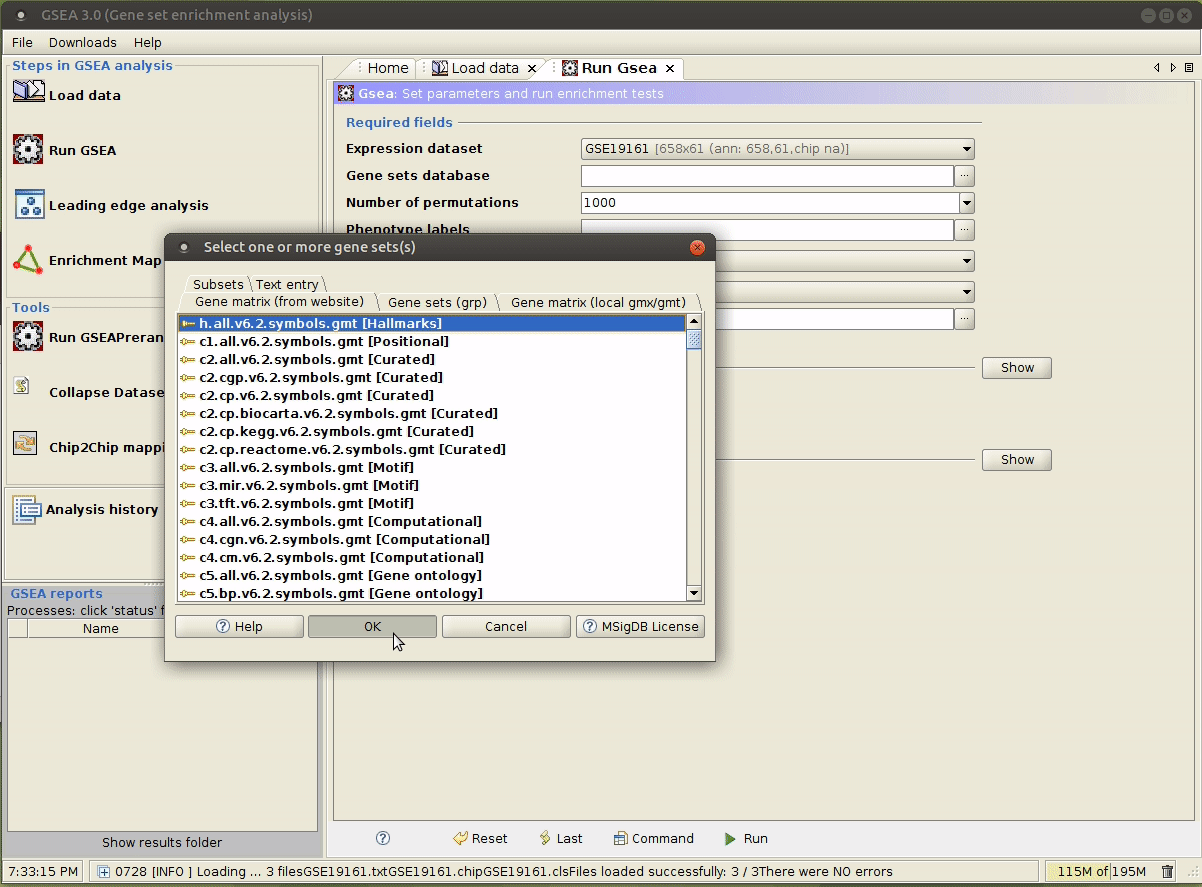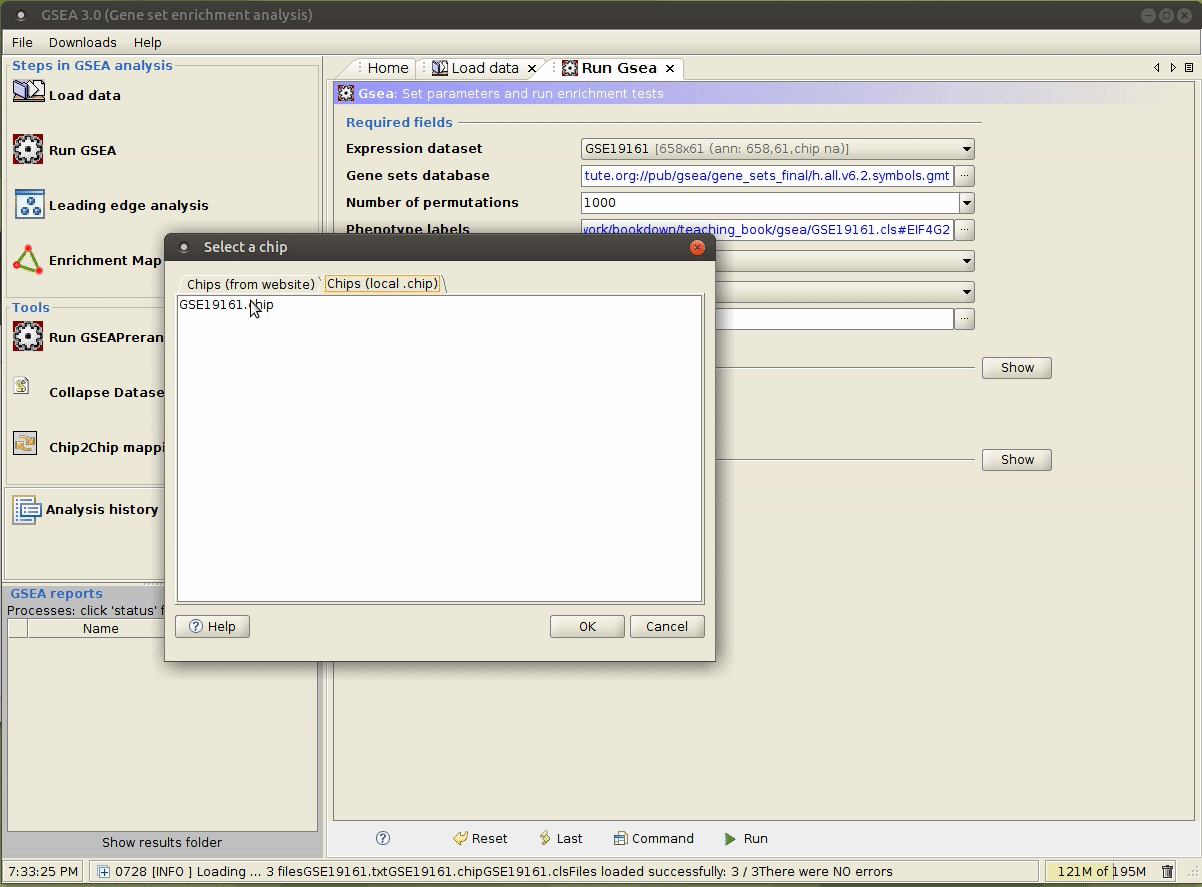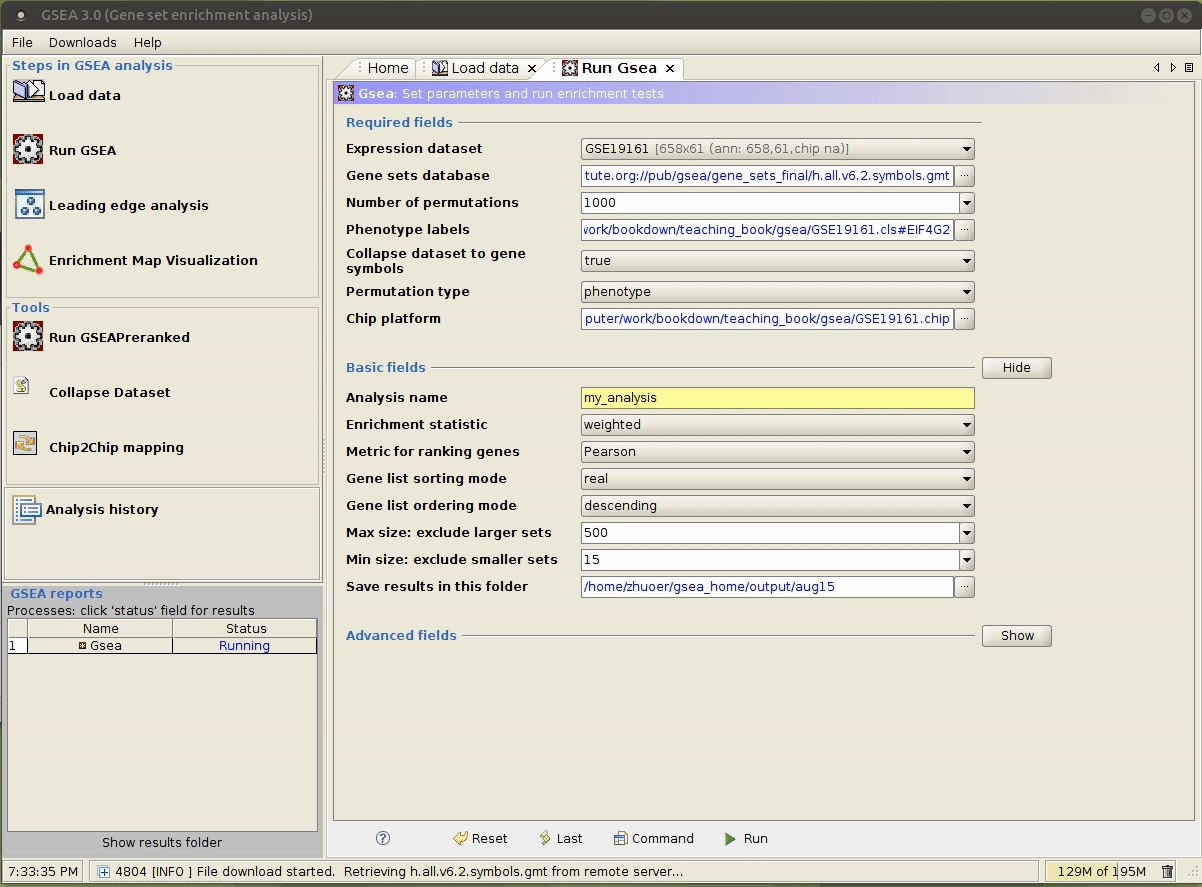
运行成功后,
Status会显示为Success,点击其即可查看输出。在本例中,没有一个 gene set 通过了显著性检验。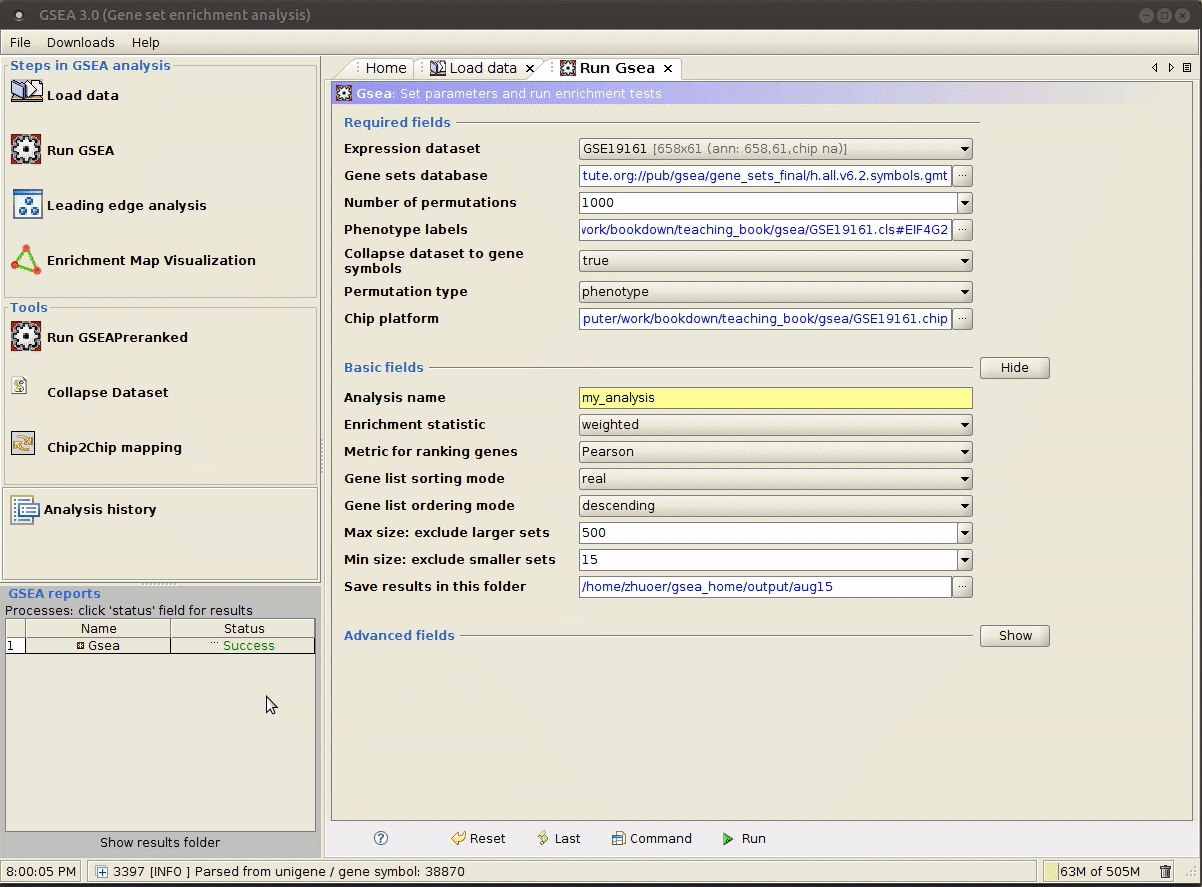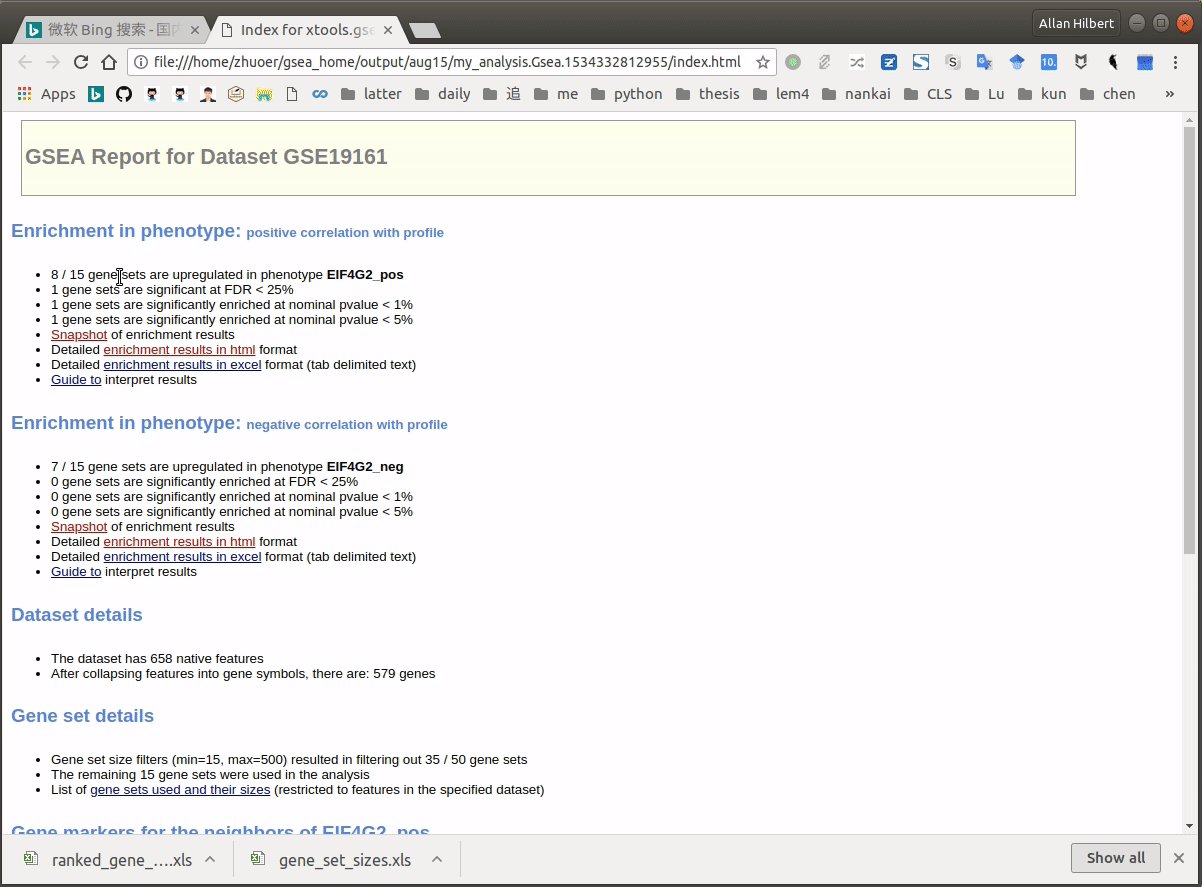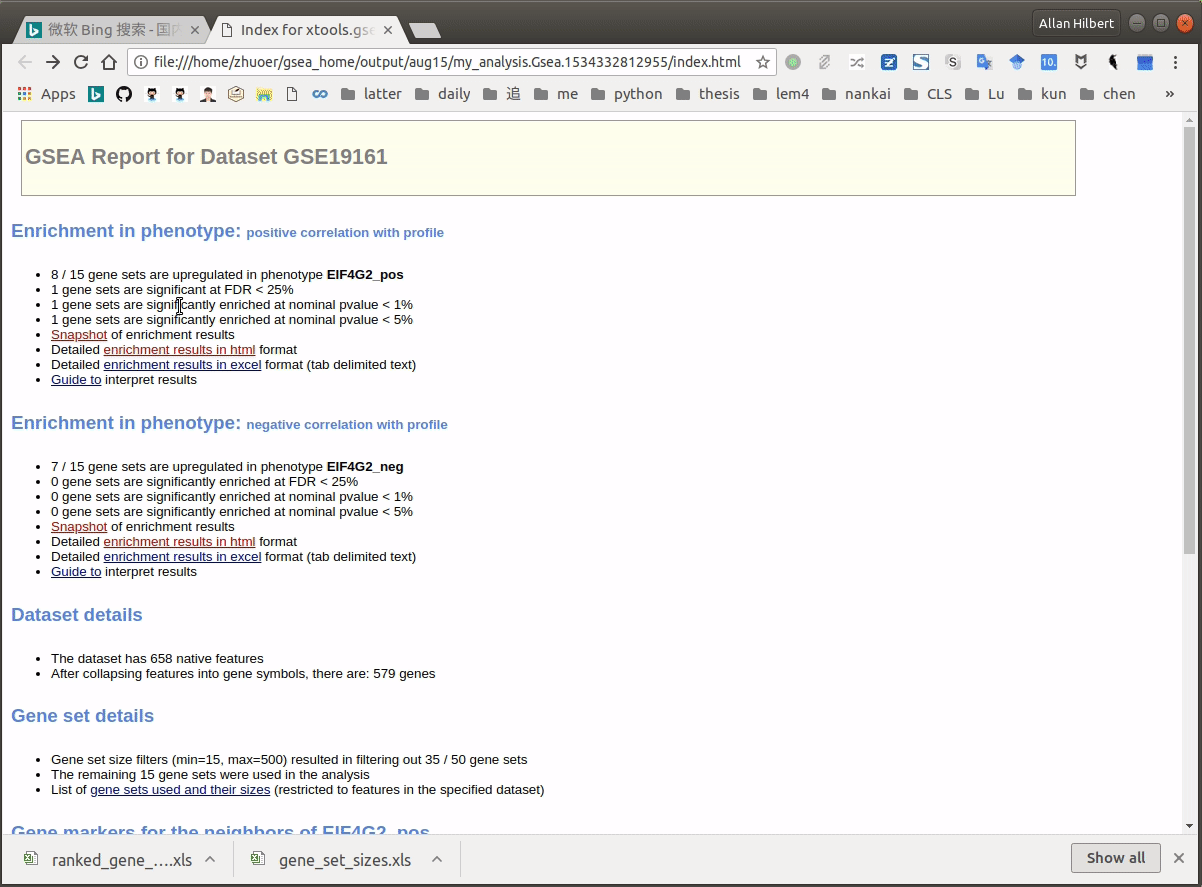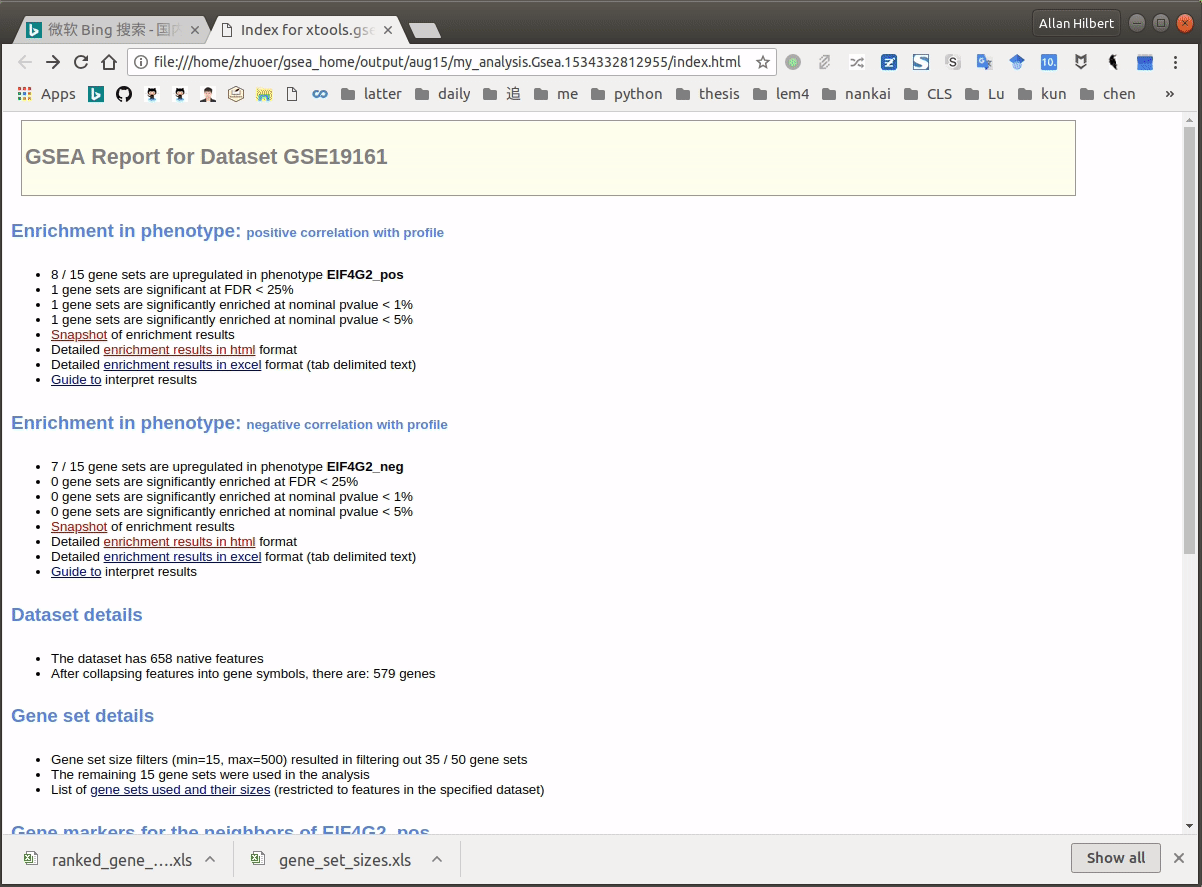
4c) 以图形化界面运行 GSEA
在上一步运行成功后,点击 Command 按钮,即可获取运行时所用的命令,如下图所示:
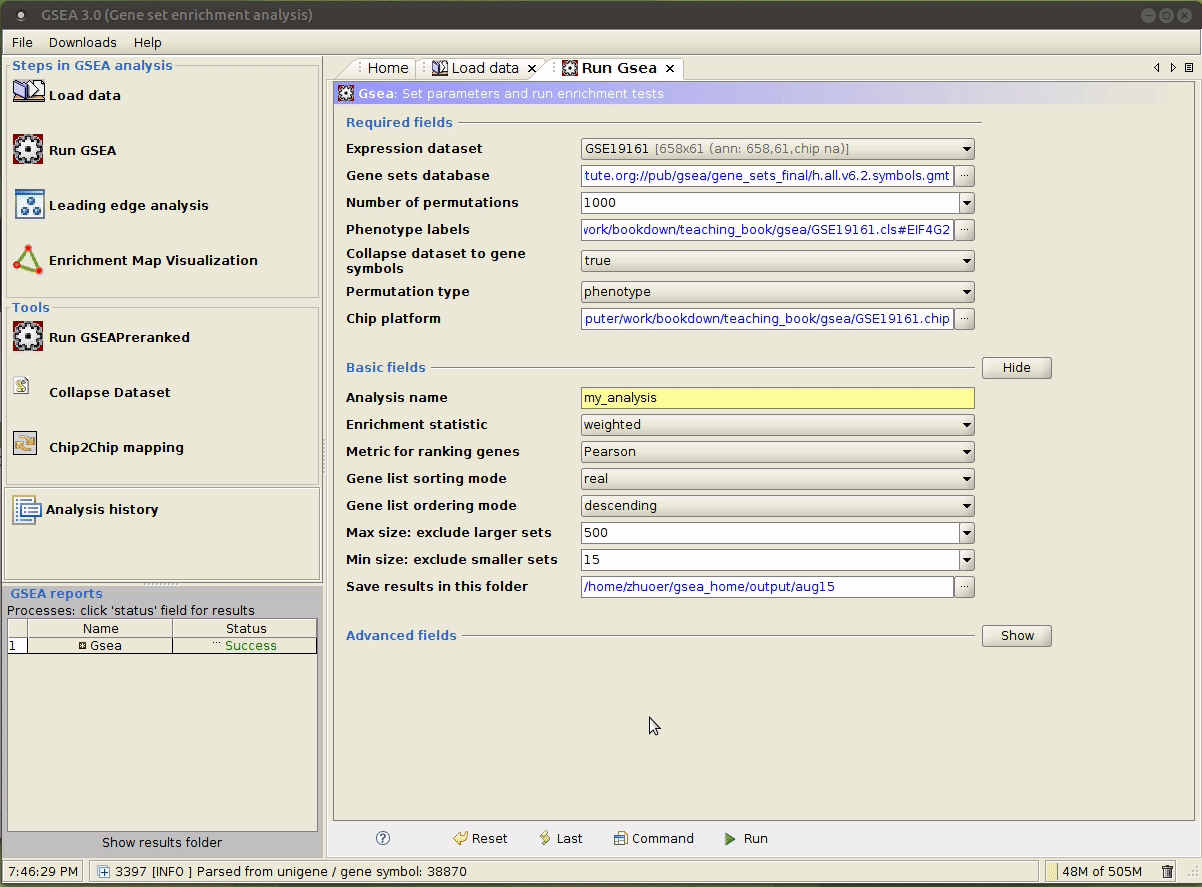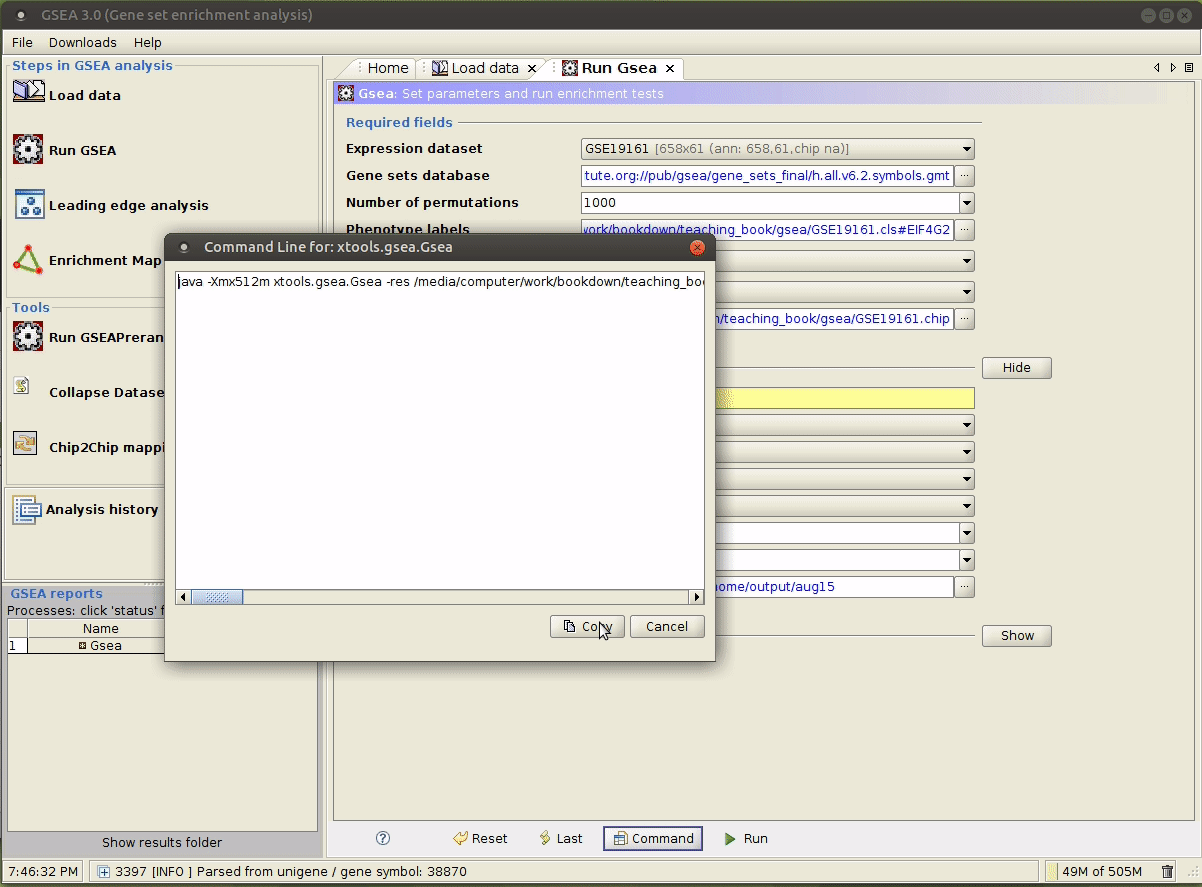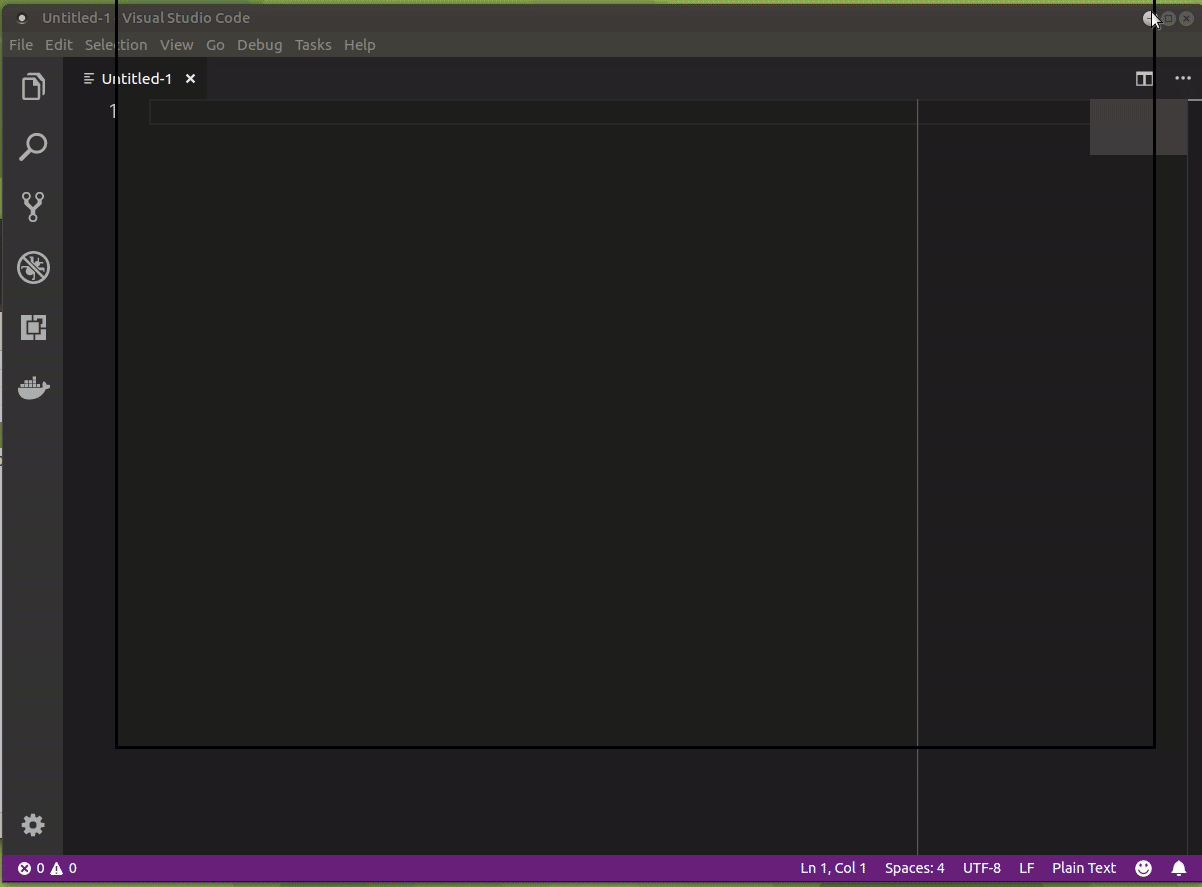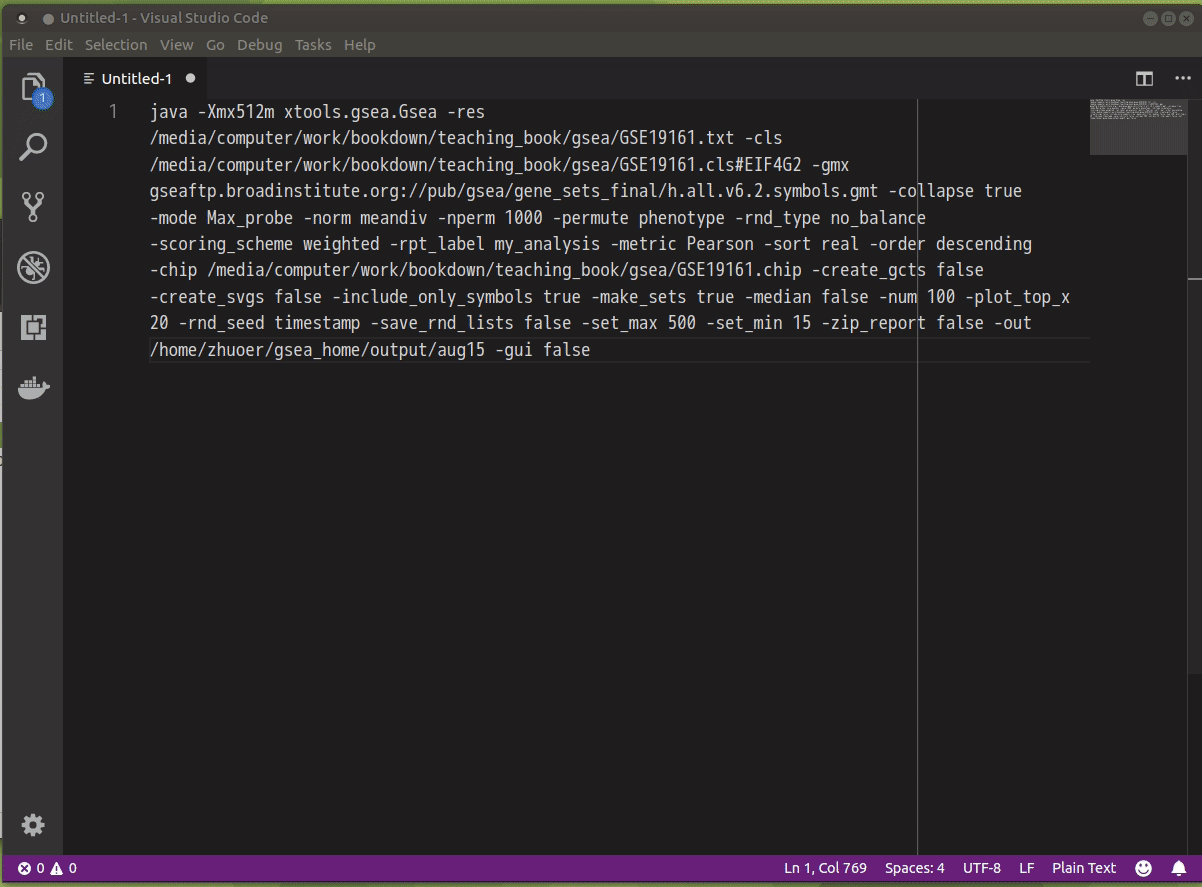
在实践中,使用默认值的参数也可以不指定,如 上文 所示。
4d) 常见错误:筛选后无基因集
由于基因芯片仅分析一部分基因,有些 gene set 会因为包含的基因过少而被剔除 。在极端情况下,可能所有的 gene set 都会被剔除,这时就会发生如下错误:

更多信息请参见http://software.broadinstitute.org/cancer/software/gsea/wiki/index.php/1001
4e) 分析离散型表型
GSEA也可以分析与离散型表型显著相关的 gene set (与差异表达相似),与上文不同的地方在于 .cls 文件。
以 GSE2251 例,打开链接后,可以在 Samples 一栏找到样品信息。本例共有12个样本,其中第1, 2, 3, 7, 8, 9 号为"E2-8",第4, 5, 6, 10, 11, 12 号为"vehicle"。针对这一表型的.cls 文件如下所示。12 是样本总数,2 是表型类型数目,0 代表第一种表型,1 代表第二种表型。(样本数过多时,网页会显示不全,这时可以解压并打开 _series_matrix.txt.gz 查看样本信息。)
12 2 1
# E2-8 vehicle
0 0 0 1 1 1 0 0 0 1 1 1
5) Homework and more
- 分析 GSE2251 中与 TP53 基因的表达量显著相关的 gene set,输入文件在 这里 下载。
注意:要求提交中间过程产生的文件(chip,cls,txt), 和最后结果,截图并解释。