6.1 Differential Expression
本章我们将:
- 理解和掌握基本的基因差异表达分析方法(Differential Expression Analysis);学会 Differential Expression Analysis 的基本软件。
- 使用 TopHat 和 Cufflinks 完成 Differential Expression Analysis。
1) Pipeline
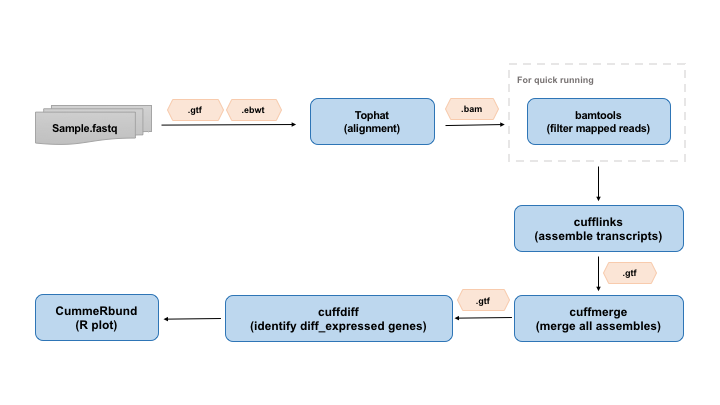
2) Data Structure
2a) getting software & data
- software (already available in Docker)
- R & cummeRbund package (BioConductor)
tophatbamtoolscufflinks
Data
The Yeast RNA-seq data were downloaded from GSE42983,有两种条件,每种两个生物重复
- wild-type :
wt1.fq&wt2.fq - H2O2 treatment (oxidative stress):
wt1X.fq&wt2X.fq
We random sample 1M or 10K reads per sample, 10K reads are in
/home/test/diff-exp/Raw_reads_10kof the Docker.- wild-type :
2b) input
| format | description | Notes |
|---|---|---|
| .fastq | RNA sequences of each sample | raw data of RNA-seq |
| .fa | sequences of the whole genome | |
| .gff | genome annotation file | Default genome annotation file is from GENCODE, is similar to gtf file |
| .ebwt | bowtie index file | used to align RNA sequences to genome |
2c) output
| format | description | Notes |
|---|---|---|
| .bam | genome mapped reads (binary format of sam) | generated by align step |
| transcript.gtf | assembled transcripts of each sample | generated by assemble step |
| merged.gtf | all annotation of assembled transcripts | generated by merge step |
| txt/tsv | differentially expressed genes | generated by DE gene identify step |
| R plot to explore differentially expressed genes | generated by R package |
cuffdiff output
| format | description |
|---|---|
| gene_exp.diff | Differentially expressed genes |
| isoform_exp.diff | Differentially expressed transcripts |
| cds_exp.diff | Differentially expressed coding sequences |
| cds.diff | Genes with differential CDS output |
| promoters.diff | Genes with differential promoter use |
| splicing.diff | Differentially spliced TSS groups |
| tss_group_exp.diff | Differentially expressed TSS groups |
3) Running Steps
首先进入到容器(在自己电脑的 Terminal 中运行,详情请参见 这里):
docker exec -it bioinfo_tsinghua bash
以下步骤均在 /home/test/diff-exp/ 下进行:
cd /home/test/diff-exp/
3a) Align the RNA-seq reads to the genome
(1) map reads using Tophat
tophat maps short sequences from spliced transcripts to whole genome.
mkdir mapping
tophat -p 4 -G yeast_annotation.gff --no-coverage-search -o mapping/wt1_thout \
bowtie_index/YeastGenome Raw_reads_10k/wt1.fq
tophat -p 4 -G yeast_annotation.gff --no-coverage-search -o mapping/wt2_thout \
bowtie_index/YeastGenome Raw_reads_10k/wt2.fq
tophat -p 4 -G yeast_annotation.gff --no-coverage-search -o mapping/wt1X_thout \
bowtie_index/YeastGenome Raw_reads_10k/wt1X.fq
tophat -p 4 -G yeast_annotation.gff --no-coverage-search -o mapping/wt2X_thout \
bowtie_index/YeastGenome Raw_reads_10k/wt2X.fq
用法:tophat [options] <bowtie_index> <reads1[,reads2,...]> [reads1[,reads2,...]]
-p/--num-threadsdefault: 1-G/--GTFGTF/GFF with known transcripts-o/--output-dirdefault:./tophat_out
(2) Extract mapped reads on chr I only (for quick running)
index
.bamfileindexcommand generates index for.bamfilebamtools index -in mapping/wt1_thout/accepted_hits.bam bamtools index -in mapping/wt2_thout/accepted_hits.bam bamtools index -in mapping/wt1X_thout/accepted_hits.bam bamtools index -in mapping/wt2X_thout/accepted_hits.bamextract
filtercommand filters.bamfiles by user-specified criteria
bamtools filter -region chrI -in mapping/wt1_thout/accepted_hits.bam \
-out mapping/wt1_thout/chrI.bam
bamtools filter -region chrI -in mapping/wt2_thout/accepted_hits.bam \
-out mapping/wt2_thout/chrI.bam
bamtools filter -region chrI -in mapping/wt1X_thout/accepted_hits.bam \
-out mapping/wt1X_thout/chrI.bam
bamtools filter -region chrI -in mapping/wt2X_thout/accepted_hits.bam \
-out mapping/wt2X_thout/chrI.bam
3b) Assemble transcripts on Chr I by Cufflinks
Assemble transcripts for each sample:
usage:
cufflinks -p number_of_cores -o output_dir input_filecufflinks -p 4 -o assembly/wt1_clout mapping/wt1_thout/chrI.bam cufflinks -p 4 -o assembly/wt2_clout mapping/wt2_thout/chrI.bam cufflinks -p 4 -o assembly/wt1X_clout mapping/wt1X_thout/chrI.bam cufflinks -p 4 -o assembly/wt2X_clout mapping/wt2X_thout/chrI.bamCreate a file called
assemblies.txtwhich lists the assembly files for each sample in theassemblyfolder. Its content is as follows:assembly/wt1X_clout/transcripts.gtf assembly/wt1_clout/transcripts.gtf assembly/wt2X_clout/transcripts.gtf assembly/wt2_clout/transcripts.gtfYou can create that file manually by
vimor use this command:ls assembly/*/transcripts.gtf > assembly/assemblies.txtMerge all assemblies to one file containing merged transcripts:
cuffmergetakes two or more Cufflinks.gtffiles and merges them into a single unified transcript catalogcuffmerge -g yeast_chrI_annotation.gff -s bowtie_index/YeastGenome.fa \ -p 4 -o assembly/merged assembly/assemblies.txt
3c) Identify differentially expressed genes and transcripts
we’ll use the output from 1M reads, not 10K reads
Run cuffdiff based on the merged transcripts and mapped reads for each sample
cuffdiff -o diff_expr -b bowtie_index/YeastGenome.fa \
-p 4 -u assembly/merged/merged.gtf \
mapping/wt1_thout/chrI.bam,mapping/wt2_thout/chrI.bam \
mapping/wt1X_thout/chrI.bam,mapping/wt2X_thout/chrI.bam
参数意义:
-o: 输出文件夹-b: Bowtie index-p: 使用的线程数-u: 转录本注释文件,一般使用 merge 得到的文件- 最后两行为每个样本的
.bam文件,这里上一行为对照组,下一行为实验组,组内的不同样本用英文逗号分隔
This won’t generate sufficient result from 10K reads。You may view the results of 1M reads in 1M_reads_diff_expr to get a better feeling of the above command.
3d) Explore differential analysis results with CummeRbund package
由于 10k reads 结果太少,我们用 1M reads 的结果(已经预先跑好)来画图
Rscript plot_DE_chart.R
图片可在 1M_reads_diff_expr 中查看。
Tips: input/output file is hard-coded in
plot_DE_chart.R, if you want to plot your own results, you need to editplot_DE_chart.R(replace1M_reads_diff_expr/with your own output directory ofcutdiff).
4) Homework
提交文件:提交差异表达的gene的 name,注明相关的fold change, p-value,FDR(q value) 等信息, 并提交绘制的 Volcano Plot。(基因列表和图最好放到一个word/pdf文件中提交)。
- 使用教程中的数据,操作时,跳过3a)(2) Extract,(直接用accepted_hits.bam 来进行 Assemble transcripts),要求找到差异表达的基因(满足 > 1, FDR < 0.05)。
- 差异表达的基因有的没有基因 name, 可以用 gene id 代替。
- 画 Volcano Plot 时,需要将所有的基因进行绘图,包括未差异表达的基因。画图用 Appenddix. Plot with R 中 7 Volcano plots 的代码画图。
回答问题:寻找 differentially expressed genes/transcripts,要对数据做怎样的处理?这些处理有哪些统计学上的考虑?
5) More Reading
- Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks Trapnell C et al. Nat Protoc. 2012
- Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown Pertea M et al. Nat Protoc. 2016